“My lab uses interdisciplinary, collaborative approaches to dissect the pathways through which cells access nutrients, leveraging this knowledge to develop innovative therapeutics that are broadly active and minimally toxic.
” – Aimee L. Edinger, VMD/PhD
SPHINGOLIPIDS AS THERAPIES FOR CANCER AND OBESITY
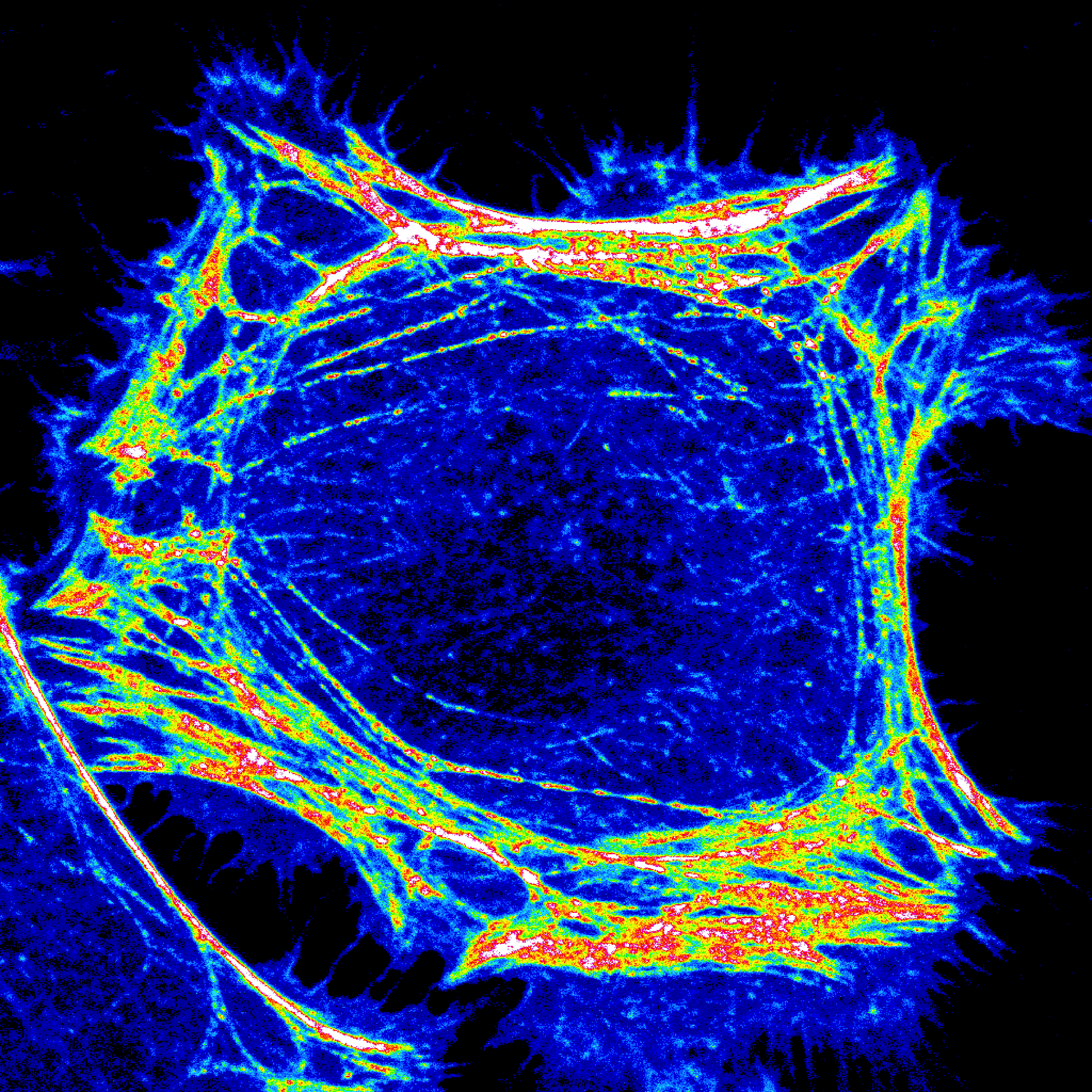
The natural product, phytosphingosine, protects yeast from stress by triggering the endocytosis and intracellular sequestration of transporters for amino acids and uracil, starving yeast into adaptive quiescence under unfavorable growth conditions. This phytosphingosine-sensitive pathway is conserved in mammalian cells where sphingosine and drug-like analogs of sphingosine produce similar effects. We have recently shown that sphingosine analogs such as SH-BC-893 (893) also control mitochondrial morphology via their effects on endolysosomal trafficking. Following consumption of a high-fat diet, palmitate accumulates in tissues where it is converted to ceramide, a toxic sphingosine derivative that triggers mitochondrial fission. Fragmented mitochondria networks produce toxic levels of ROS and lead to ER stress that causes cellular dysfunction. Synthetic sphingosine analogs prevent the recruitment of the mitochondrial fission factor DRP1, preserving tubulated mitochondrial networks, preventing excessive ROS generation and ER stress. In mice with diet-induced obesity, SH-BC-893 restores normal weight and metabolic function. These orally bioavailable synthetic sphingolipids block mitochondrial fission more robustly than any other small molecule or peptide inhibitor described to date. Because of these mitochondrial effects, SH-BC-893 and related compounds may be useful in other lethal diseases that stem from excessive mitochondrial fission: cardiovascular disease, type II diabetes, nonalcoholic steatohepatitis, chronic kidney disease, neurodegenerative disease, and cancer drug resistance. By activating PP2A, SH-BC-893 and other sphingosine analogs also control the nuclear translocation of important transcription factors, currently an area of active investigation in the lab.
DRUG DEVELOPMENT
Through a long-term collaboration with Stephen Hanessian’s chemistry group, we have dissected the structural features of SH-BC-893 that are critical for its ability to disrupt intracellular trafficking. A series of analogs have been developed that are differentially active, producing some, but not all, of the 893-associated phenotypes. These compounds are powerful tools to dissect the pleiotropic actions of sphingolipids. Working with Pierre Thibault’s proteomics group at the University of Montreal, we have identified the protein targets of SH-BC-893 and other sphingosine analogs. We are now using biochemical assays, molecular modeling, and crystal structures to map the specific binding sites of 893 and related natural sphingolipids on these protein targets. Structure-driven drug design efforts currently underway will lead to new analogs with increased potency and selectivity.
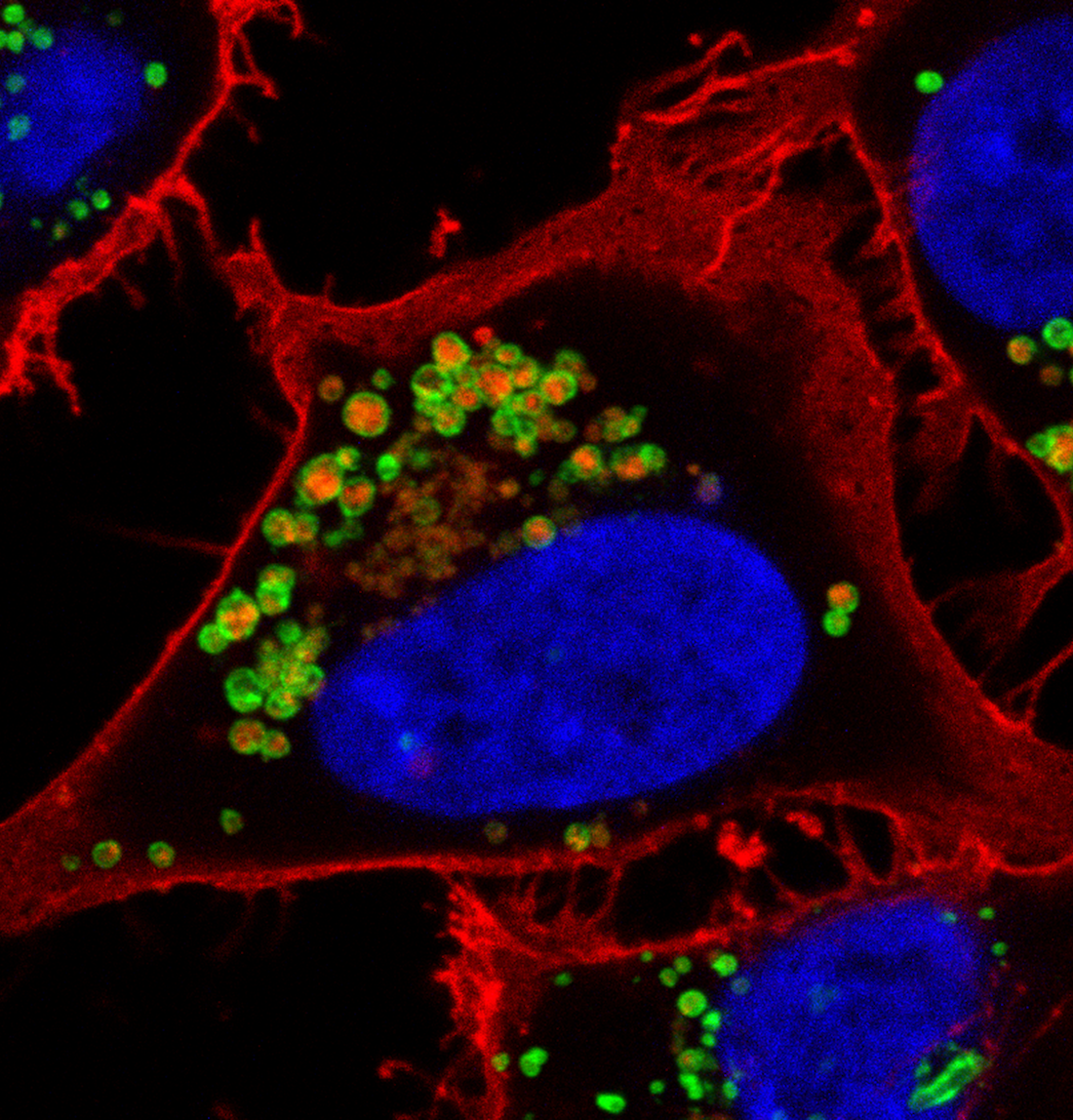
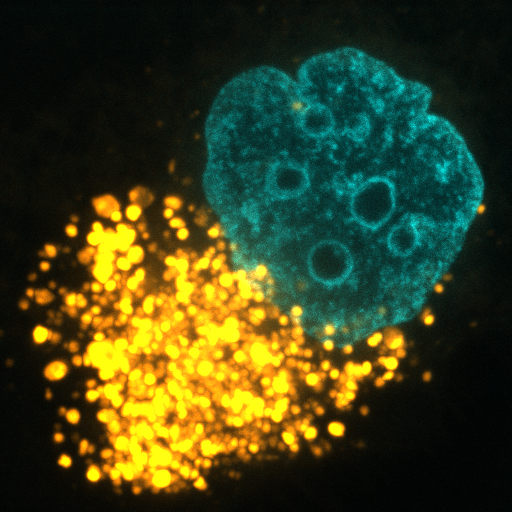
MACROPINOCYTOSIS AND NUTRIENT SCAVENGING
Cancer cells require a steady stream of nutrients to support their unchecked growth. However, the blood vessels that supply these nutrients are often inadequate, tortuous, and leaky. Desmoplasia can also lead to elevated interstitial pressure that collapses tumor blood vessels. Cancer cells overcome these limitations on nutrient delivery by scavenging macromolecules from the microenvironment. One scavenging strategy employed by tumors is macropinocytosis, a process by which extracellular material is non-specifically engulfed. Oncogenic mutations in KRAS and PI3K and loss of function mutations in PTEN drive macropinocytosis in pancreatic, breast, and prostate cancer. In each of these tumor classes, macropinocytosis confers the ability to proliferate in nutrient-limiting conditions. Blocking macropinocytosis significantly inhibits tumor growth, even causing tumor regressions. We have identified AMP-activated protein kinase (AMPK) as a critical regulator of macropinocytosis downstream from oncogene activation. In addition, we have shown that macropinocytic tumor cells can feed off the corpses of their dead neighbors through a process we termed “necrocytosis.” The conditions present in virtually all solid tumors will favor nutrient scavenging and, because both KRAS activation and PI3K pathway activation can drive macropinocytosis, macropinocytosis inhibitors have the potential to make a large therapeutic impact across cancer classes. On-going research in the Edinger lab address key open questions in the field: How pervasive is macropinocytosis across different tumor types? Can we target macropinocytosis to limit drug resistance? How do agents that interfere with macropinocytosis affect the tumor microenvironment? Answering these questions will provide novel insights into tumor cell biology and could provide a rationale for new therapeutic strategies.
NUCLEOTIDE THERAPEUTIC DELIVERY
Multiple antisense oligonucleotide (ASO) and siRNA therapies are now FDA-approved for chronic diseases demonstrating the feasibility and safety of this strategy for reducing the level of cellular RNAs. ASO and siRNA are the ultimate platform technology. In principle, any RNA, coding or non-coding, can be targeted making the “undruggable” targetable and the applications nearly limitless. Medicinal chemistry optimization over the last three decades has solved historical problems with ASO stability and rapid clearance. However, a barrier to the widespread application of ASO therapeutics remains: inefficient delivery to the intracellular RNA targets. The majority of the systemically delivered ASO is not taken up by cells, and much of what is endocytosed ends up sequestered in the lysosome where ASO is stable, but inactive. Our goal is to use our knowledge of endolysosomal trafficking to identify small molecules that enhance ASO activity in normal tissues other than the liver and in tumors. Enhancing ASO activity in tumors would allow us to target “undruggable” oncogenes. If these compounds increase ASO activity in normal, extrahepatic cells, they could dramatically expand the number of diseases that can be treated with antisense therapies.